|






|
|
History of polyketide
research
Polyketides as a research area dates back to 1893, where
Collie and Myers isolated the first polyketide; orcinol (Collie & Myers
1893). Later the same year Collie proposed a rough mechanism for the
synthesis of orcinol and related compounds, based solely on their structure.
In this, he states that they most likely are synthesized by repetitive
condensation or polymerization reactions (Collie 1893). Collie later showed
that orcinol and related compounds did not fit into any of the previously
described chemical classes, and therefore proposed that they should be
placed in a new class called polyketides. Its members being characterized by
the repetitive occurrence of a -CH2-CO- “motif”, which he named
ketide (Bentley & Bennett 1999).
For the following 45 years, only chemists explored the
world of polyketides, and many new compounds were isolated and chemically
characterized from both filamentous fungi and eubacteria (Streptomyces
sp.). Research in the biosynthesis mechanisms was initiated in 1953,
when Birch and Donovan suggested a new biosynthesis pathway for polyketides,
which in many aspects resembled the mechanism suggested for fatty acid
biosynthesis. This hypothesis became known as the polyacetate hypothesis and
stated that: “Polyketides are formed by the head-to-tail linkage of acetate
units, followed by a cyclization by an aldol reaction or by acylation to
phenols” (Birch & Donovan 1953). In successive studies with the newly
developed radionuclear labelling technique, it was possible to show that the
ketide groups found in polyketides originated from acetate units derived
from the primary metabolism of the producing organism. This theory has
proven extremely successful in explaining the biochemical relationship
between the different isolated derivates from wild types and deletion
mutants, incapable of producing the original polyketide in question (Bentley
& Bennett 1999).
With the implementation of modern recombinant DNA
techniques (Ligase (1967), Restriction enzymes (1970), recombinant DNA
(1972), sequencing (1983) and PCR (1984) (Kielberg et al. 2003)) in
the mid 1980’s, it became possible to analyse the genetic basis for the
production of polyketides. The first polyketide to be understood in genetic
and molecular biological terms was the blue pigment actinorhodin produced by
Streptomyces coelicolor. Early classical genetic analysis of six
classes of act mutants had shown that they were closely linked, and
by recombinant DNA technology it was possible to clone a single DNA piece
that could compensate for all six known classes of mutants (Rudd & Hopwood
1979) (Hopwood 1997). The genes responsible for the production of a single
polyketide (and other secondary metabolites) are typically organized in
clusters of tightly linked genes (operons in bacteria and true gene
clusters in fungi).
Polyketide synthases
Polyketide synthases (PKSs) are structurally and
functionally related to fatty acid synthases (FAS’s), as both enzyme classes
catalyzes the condensation of activated primary metabolites (acetyl-CoA and
malonyl-CoA) to form
b-ketoacetyl
polymers linked to the enzyme by thioester bonds.
CO2-CH2-CO-S-CoA
+ CH3-CO-S-PKS => CH3-CO-CH2-CO-S-PKS
+ CoA-H + CO2
In the fatty acid
synthesis, this condensation is followed by
b-ketoreduction,
dehydration and enoyl reduction to yield the final fully reduced (saturated)
fatty acid. In polyketide synthesis these reduction steps are partly or
completely omitted in a controlled fashion, resulting in a highly diverse
polyketide chain with respect to the occurrence of
b-ketone,
b-hydroxyl
and alkyl groups (Fujii et al. 2001).
Polyketide synthases (PKSs) has typically been
categorized based on their number of subunits (a single or multiple) and
mode of synthesis (linear or iterative) (Table 2).
Eleven different catalytic domains is generally recognized in PKSs (Table
3). The simplest functional PKS consists of a KS, an AT, an ACP and a TE
domain. The domains responsible for the addition of a single ketide unit to
the growing polyketide and the following modification is denoted a module (http://www.nii.res.in/nrps-pks.html)
(Fujii et al. 2001).
The best characterized class of PKSs is the type I
modular, but the functional information derived from these typically
also apply to the other classes of PKSs.
|
Group |
Protein structure |
Synthesis mechanism |
Predictable |
Resembles |
Found in |
|
Type I (modular) |
Single protein with multiple modules. |
Linear (assembly-line style) in which each active
site is used only once. |
Yes, to some extent. |
na |
Bacteria |
|
Type I (iterative) |
Single protein with one module. |
Iterative, in which the active sites are reused
repeatedly. |
No |
Vertebrate FAS |
Fungi |
|
Type II |
Multiple proteins, each with a single mono-functional
active site. |
Iterative, in which active sites may be used only
once or repeatedly. |
No |
Bacterial FAS |
Bacteria |
|
Type III |
Single
protein with multiple modules |
Iterative, in which the active sites are reused
repeatedly. |
No |
na |
Plants and Bacteria |
Table 2
The differences between the three types of PKSs with respect to structure,
synthesis mechanism, evolutionary relation and distribution (Watanabe &
Ebizuka 2004).
|
Active site |
|
Function |
|
Starter Acyltransferase (SAT) |
C |
Loading of stater units |
|
Acyltransferase (AT) |
C |
Loading of starter, extender and intermediate acyl
units. |
|
Acyl Carrier protein (ACP) |
C |
Holds the growing polyketide chain as a thiol ester
(KS-S-polyketide). |
|
b-ketoacyl
synthase (KS) |
C |
Condensation reaction between starter/intermediate
and extender units. |
|
b-keto reductase (KR) |
R |
Reduces
b-ketone groups to hydroxyl groups. |
|
Dehydratase (DH) |
R |
Reduces hydroxyl groups to enoyl groups
(unsaturated). |
|
Enoyl reductase (ER) |
R |
Reduces enoyl groups to alkyl groups (saturated). |
|
Thioesterase (TE) |
C |
Facilitates the release of the final product from the
enzyme. |
|
Methyltransferase (MT) |
M |
Transfers methyl groups to the growing polyketide. |
|
Product template domain |
C |
Determines the folding pattern of the
polyketide backbone in non-reducing iPKSs |
|
Claisen cyclase (CYC) |
M |
Facilitates ring formation by a Claisen-type
cyclization reaction. |
|
Condensation (CON) |
M |
Facilitates the condensation of the synthesized
polyketide with other polyketides. |
Table 3
The different types of domains found in PKSs. The eleven different domains
can be divided into three groups based on which part of the synthesis they
participates in (C = condensation reaction, R = reduction of
b-ketone and M
= other post-condensation modifications).
Type I (modular) PKSs
The final number of ketide units in polyketides
synthesized by type I (modular) PKS equals the number of modules found in
the PKS. This is a result of the linear synthesis mode of these PKSs, where
the growing intermediate is passed along the PKS from module to module
(Figure 3). The TE domain mediates the release of the final polyketide.
This type of PKSs is found in bacteria and is
responsible for synthesis of clinical and economical important macrolide
polyketides, such as the erythromycin A and rifamycin (http://www.bio.cam.ac.uk/~pflgroup/research.htm).
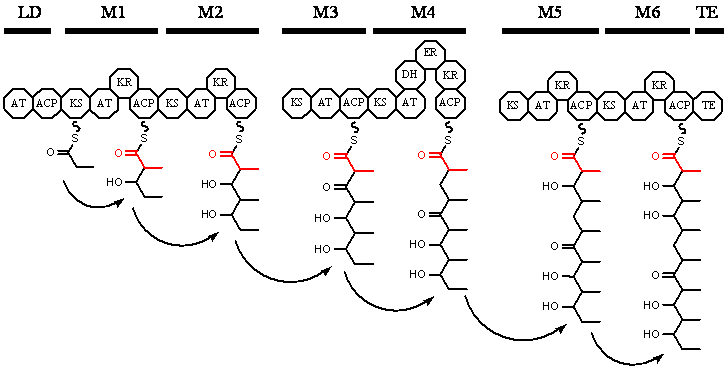
Figure 3
The module structure of the three Type I (modular) PKSs responsible for the
synthesis of erythromycin, with the growing polyketide chain shown, the
newest ketide group in each step is highlighted with red. The thick lines at
the top denote the extent of the individual module, note that erythromycin
is synthesized by three separate enzymes consisting of two modules each
(After
http://www.bio.cam.ac.uk/~pflgroup/research.htm).
Abbreviations for the domains and their function can be found in Table 3. LD
= loading module, M1-M6 modules and TE = Thioesterase domain.
Polyketide synthases and prediction
of their products
Polyketide synthases are known from both eukaryotic and prokaryotic systems.
This family of enzymes catalyze the fusion of short carbon chains into long
polymers, via successive rounds of Claisen condensation reactions. However,
while catalyzing similar reactions there are several different classes of
PKSs, differing in their domain architecture and mode of synthesis.
Type I PKS are characterized by being multizymes (A single polypeptide chain
housing multiple different active sites capable of catalyzing different
reactions) posing all the necessary enzymatic domains for the formation of a
polyketide. In type II PKS the required catalytic domains are located on
individual proteins that interact to form a functional PKS enzyme complex.
The type III PKS (chalcone synthase-like) differ from the two other types by
not relying on acyl carrier protein domains (Meier and Burkart, 2009).
The type I PKS can furthermore be subdivided into a modular and an iterative.
The modular type poses multiple copies of each type of active site,
organized into modules that are responsible for the addition and
modification of a single ketide unit. The starter unit is loaded into the
enzyme in an N’terminal loading domain. The growing polyketide chain is then
passed from module to module until it reaches the C’terminal end of the
enzyme where it is released by a thioesterase domain. This means that it is
possible to predict the final polyketide length and which types of
modifications the individual ketides units will harbour, just by deciphering
the order of catalytic sites found along the PKS and the number of modules
(Meier and Burkart, 2009). The iterative type of PKS (iPKS) only pose a
single copy of each catalytic domain, however these can be deployed
repeatedly during synthesis of a single polyketide molecule, as described in
the next two sections. Type I iPKSs are typically further subdivided based on
which modifications they can introduce into the growing polyketide chain
during synthesis. However the action of the core domains (also known as the
minimal PKS) remain the same in all subclasses.
Minimal iPKS: Action of the core domains (non-reducing PKSs)
Polyketide biosynthesis in many aspects
resembles the fatty acid synthesis, by utilizing the same active sites and
reaction mechanisms. The synthesis can be divided into several steps, shown
in figure 5. First the starter unit, in the form of an acetyl, is loaded
into the
b-ketosynthase domain (KS) of the enzyme, a process that is
mediated by the acyl-carrier-protein domain (ACP) (step 1 in figure 5). The
acetyl is delivered to the enzyme in the form of acetyl-CoA and bound to the
enzyme via a thioester bond (Nelson and Cox, 2005). ACP domains in
non-reducing iPKSs have been shown to be able to auto-malonylate (Hitchman
et al., 1998). The ACP domain includes a long flexible prosthetic group
(4’-phosphopatetheine) that functions as a “crane” that moves the substrates,
intermediates and products between the different active sites found in the
iPKS (Nelson and Cox, 2005).
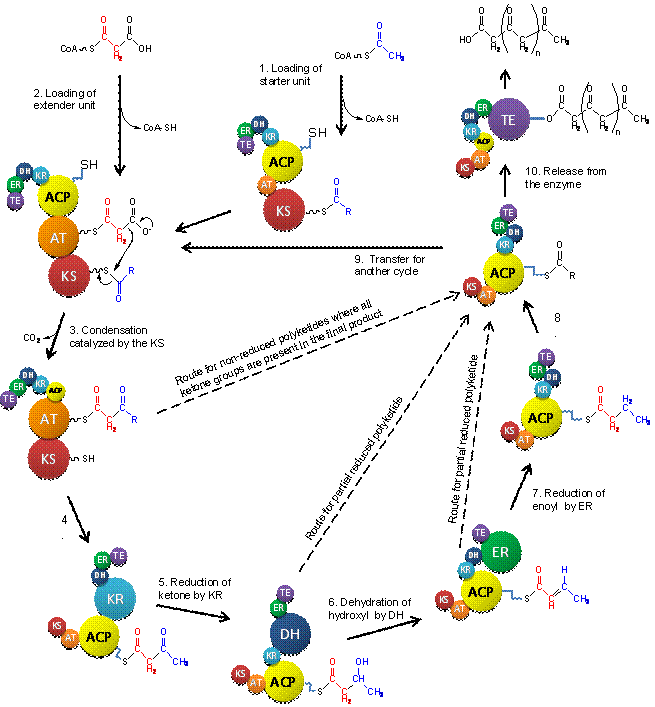

The second substrate for the Claisen
condensation reaction, the extender unit, is then loaded into the AT domain
by the Malonyl-CoA domain (MAT) (step 2 in figure 5). The MAT is only found
in the non-reducing iPKS where it facilitates the loading of malonyl between
CoA and the acyl-transferase (AT) domain. The extender unit is typically a
malonyl delivered to the enzyme by CoA. The KS domain catalyze the Claisen
condensation reaction between the starter and extender units, driven by
decarboxylation of the extender unit (Proctor et al., 1999) (Step 3 in
figure 5). At this point two different options exist: 1) add another ketide
unit or 2) release the polyketide chain from the enzyme. In option 1 the
product is transferred back to the KS domain to prepare for a second
iteration (step 9 in figure 5) and another extender unit is loaded into the
enzyme. For option 2 to occur the polyketide have to have reached its
predetermined length which is unique for each iPKS. The polyketide is
transferred to the thioesterase domain (TE) that catalyzes its release from
the enzyme (Hendrickson et al., 1999) (Step 10 in figure 5). The products of
non-reducing PKSs typically undergo intra-chain aldol or Claisen reactions
catalyzed by a Claisen-type cyclase domain (CLC), which is related to the
TE, resulting in the formation of aromatic structures (Fujii et al., 2001).
Recent results have proven the existence of a product template domain (PT)
that is responsible for situating the polyketide chain correctly to ensure
that only one product type is formed (Crawford et al., 2009).
Mammalian fatty acid synthases have been shown to function as homodimers (head-to-head
arrangement), meaning that they pose two copies of each domain. Experiments
with heterodimers, where one of the momomers have been mutated, suggest that
the two monomers feed each other substrates. It is likely that a similar
situation exist for the iPKS, but it has not been experimentally validated (Witkowski
et al., 2004).
Action of modifying domains
The ketide units that are added to the growing polyketide chain by the
core set of PKS domains (KS, AT and ACP) can be subjected to modifications
catalyzed by modifying domains, if such are present in the enzyme. In
reducing iPKSs the ketone group of a ketide unit can be reduced to various
degrees catalysed by ketoreductase (KR), dehydratase (DH) and enoyl
reductase (ER) domains (Kroken et al., 2003). The KR domain is responsible
for reducing the ketone group to a hydroxyl group (Step 5 in figure 5), the
DH domain further reduces the hydroxyl group to an enoyl group (Step 6 in
figure 5), which in turn can be reduced to an alkyl group catalyzed by the
ER domain (Step 7 in figure 5). Reducing iPKSs can also contain domains that
add methyl (CmeT) or acetyl (CacT) groups to the reduced polyketide chains,
resulting in branching of the backbone chain (Song et al., 2004).
Subdivision of type 1 iPKSs
Fungal iterative Polyketide
synthases have traditionally been divided into three groups based on their
modifying domains: non-reducing iPKS (NR iPKS), partial reducing iPKS (PR
iPKS) and fully reducing or highly reducing iPKS (FR iPKS). The NR iPKS are
characterized by only having the core set of iPKS domains (AT, KS, ACP and
TE/CYC) as well as MAT and PT domains. The PR iPKS contain, in addition to
the core set of domains, a KR domain and possibly also a DH resulting in
products with hydroxyl and enoyl groups. The HR iPKS contains all the
domains needed to reduce the
b-ketone group to an alkyl group, meaning that
they in addition to the core domains also have KR, DH and ER domains.
However it is important to note that the presence of a modifying domain does
not necessarily mean that it is used in every iteration of synthesis, a good
example of this is found in the fusarin C biosynthetic pathway. This
classification system reflects the evolutionary history of the iPKS as
proven by the analysis of KS domains made by (Kroken et al., 2003). This is
however a very rough classification that does not encompass the complex
nature of iPKS biosynthetic systems, especially if one includes the
different types of possible hybrid enzymes and multienzyme systems: such as
systems where two iPKSs interact to form a common product (see
zearalenone),
or where an iPKS and a NRPS have been fused to form a single enzyme (see
fusarin C),
or systems where iPKS products are modified by NRPS like enzymes (see
fumonisins).
Animation of a minimal type I Iterative PKS in
action (PowerPoint created by Rasmus J.N. Frandsen)
(click to download)
References
 |
Meier,J.L.,
and Burkart,M.D. (2009) The chemical biology of modular biosynthetic
enzymes. Chemical Society Reviews 38: 2012-2045. |
|
Nelson,D.L., and Cox,M.M. (2005) Lipid Biosynthesis in Lehninger
Principles of biochemistry, fourth edition. New
York, USA : W.H. Freeman and Company. |
|
Proctor,R.H., Desjardins,A.E., Plattner,R.D., and Hohn,T.M. (1999) A
polyketide synthase gene required for
biosynthesis of fumonisin mycotoxins in Gibberella fujikuroi slating
population A. Fungal Genetics and Biology 27:
100-112. |
|
Hendrickson,L., Davis,C.R., Roach,C., Nguyen,D.K., Aldrich,T., Mcada,P.C.,
and Reeves,C.D. (1999) Lavastatin biosynthesis
in Aspergillus terreus: characterization of blocked mutants, enzyme
activities and a multifunctional polyketide
synthase gene. Chemistry & Biology 6: 429-439. |
|
Fujii,I.,
Watanabe,A., Sankawa,U., and Ebizuka,Y. (2001) Identification of Claisen
cyclase domain in fungal polyketide synthase WA,
a naphthopyrone synthase of Aspergillus nidulans. Chemistry & Biology 8:
189-197. |
|
Crawford,J.M., Korman,T.P., Labonte,J.W., Vagstad,A.L., Hill,E.A.,
Kamari-Bidkorpeh,O. et al. (2009) Structural
basis for biosynthetic programming of fungal aromatic polyketide
cyclization. Nature 461: 1139-1143. |
|
Witkowski,A., Ghosal,A., Joshi,A.K., Witkowska,H.E., Asturias,F.J., and
Smith,S. (2004) Head-to-head coiled arrangement
of the subunits of the animal fatty acid synthase. Chemistry & Biology 11:
1667-1676. |
|
Kroken,S.,
Glass,N.L., Taylor,J.W., Yoder,O.C., and Turgeon,B.G. (2003) Phylogenomic
analysis of type I polyketide synthase genes in
pathogenic and saprobic ascomycetes. Proceedings of the National Academy
of Sciences of the United States of America 100:
15670-15675. |
|
Song,Z.S.,
Cox,R.J., Lazarus,C.M., and Simpson,T.J. (2004) Fusarin C biosynthesis in
Fusarium moniliforme and Fusarium venenatum.
Chembiochem 5: 1196-1203. |
|
Bentley R.
and Bennet J.W. “Construction Polyketides: From Collie to Combinatorial
Biosynthesis”, Annual Review of Microbiology (1999) Vol. 53, p. 411-446 |
|
Bingle L.E.,
Simpson T.J. and Lazarus C.M. “Ketosynthase domain probes identify two
subclasses of fungal polyketide synthase genes”, Fungal Genetics and
Biology (1999) Vol. 26, No. 3, p. 209-223 |
|
Birch A.J.
and Donovan F.W. “Studies in relation to biosynthesis. I. Some possible
routes to derivatives of orcinol and phloroglucinol”, Australian Journal
of Chemistry (1953) Vol. 6, p. 360-368 |
|
Chasseur
C., Suetens C., Michel V., Mathieu F., Begaux F., Nolard N. and Haubruge
E., “A 4-year study of the mycological aspects of Kashin-Beck disease in
Tibet”, International Orthopaedics (2001), Vol. 25, No. 3, p. 154-158. |
|
Collie J.N.
“The production of naphthalene derivatives from dehydracetic acid”,
Journal of the Chemical Society (1893), Vol. 63 p. 329-337 |
|
Department
of crop sciences University of Illinois at Urbana-Champaign “Mycotoxins
and mycotoxicoses”, Reports on Plant Diseases No. 1105 (August 1997) |
|
Desjardins
A.E. “Gibberella from A (venaceae) to Z (eae)”, Annual review of
Phytopathology (2003), Vol. 41, p. 177-198
Dvorska J.E. “Effect of aurofusarin, a mycotoxin produced by Fusarium
graminearum, on Japanese quails” Abstract of International Symposium
Bioactive Fungal metabolites – Impact and Exploitation (2001), p. 32-33 |
|
Dvorska J.E.,
Surai P.F., Speake B.K. and Sparks N.H. “ Protective effect of modified
glucomannans against aurofusarin-induced changes in quail egg and embryo”,
Comparative biochemistry and physiology. Toxicology & pharmacology (2003),
Vol. 135C, p. 337-343 |
|
Forsyth D.M.,
Yoshizawa T., Morooka N. and Tuite J. “Emetic and Refusal Activity of
Deoxynivalenol to Swine”, Applied and Environmental Microbiology (1977),
Vol. 34, No. 5, p. 547-552 |
|
Fujii I.,
Watanabe A., Sankawa U. and Ebizuka Y. “Identification of Claisen cyclase
domain in fungal polyketide synthase WA, a naphthopyrone synthase of
Aspergillus nidulans”, Chemistry & Biology (2001), Vol. 8, p. 189-197 |
|
Gokhale R.S.,
Tsuji S.Y., Cane D.E. and Khosla C. “Dissecting and Exploiting
Intermodular Communication in Polyketide Synthases”, Science (2000), Vol.
284, No. 5413, p. 482-485 |
|
Hendrickson
L., Davis C.R., Roach C., Nguyen D.K., Aldrich T., McAda P.C. and Reeves
C.D. “Lovastatin biosynthesis in Aspergillus terreus: characterization of
blocked mutants, enzyme activities and a multifunctional polyketide
synthase gene”, Chemical Biology (1999) Vol. 6, No. 7, p. 429-439 |
|
Hitchman
T.S., Crosby J., Byrom K.J., Cox R.J. and Simpson T.J. “Catalytic self-acylation
of type II polyketide synthase acyl carrier proteins”, Chemistry & Biology
(1998), Vol. 5, No. 1, p. 35-47 |
|
Hopwood
D.A. “Genetic Contributions to Understanding Polyketide Synthases”,
Chemical Reviews (1997), Vol. 97, No. 7, p. 2465-2498 |
|
Kielberg
V., Nørby S. and Rasmussen L. “DNA og RNA – en håndbog” printed in
Copenhagen, DK, Gads Forlag (2003)
Madigan M.T., Martinko J.M. and Parker J. “Brock Biology of Microorganisms,
9th edition” Prentice Hall Inc. New Jersey USA 2000, p. 387 Deacon J.W.
“Modern Mycology, 3th edition” printed in Cambridge UK (1997) |
|
Malz S.,
Grell M. N., Thrane C., Maier F. J., Rosager P., Felk A., Albertsen K.S.,
Salomon S., Bohn L., Schäfer W. and Giese H. “Identification of a gene
cluster responsible for the biosynthesis of aurofusarin in the Fusarium
graminearum species complex”, Fungal Genetics and Biology (2005), (article
in press). |
|
Marasas
W.F., Kellerman T.S., Gelderblom W.C., Coetzer J.A., Thiel P.G. and van
der Lugt J.J., “Leukoencephalomalacia in a horse induced by fumonisin B1
isolated from Fusarium moniliforme”, Onderstepoort Journal of Veterinary
Research (1988), Vol. 55, No. 4, p. 197-203 |
|
Marasas
W.F.O., Nelson P.E. and Toussoun T.A. “Toxigenic Fusarium species:
Identity and mycotoxicology” from Pennsylvania State University Press, USA
(1984) |
|
Medentsev
A.G and Akimenko V.K. “Naphthoquinone metabolites of the fungi”,
Phytochemistry (1998), Vol. 47, No.6 p. 935-959 |
|
Nelson D.L.
and Cox M.M. “Lehninger Principles of biochemistry, fourth edition”
printed in New York, USA (2005) |
|
Nelson P.
E., Dignani M. C. and Anaissie E. J. “Taxonomy, Biology, and Clinical
Aspects of Fusarium Species”, Clinical Microbiology Review (1994), Vol. 7,
No. 4, p. 479-504 |
|
Proctor R.H.,
Desjardins A.E., Plattner R.D. and Hohn T.M. “A Polyketide Synthases Gene
Required for Biosynthesis of Fumonisin Mycotoxin in Gibberella fujikuroi
Mating Population A”, Fungal Genetics and Biology (1999), Vol. 27, p.
100-112 |
|
Rudd B.A.M.
and Hopwood D.A. “Genetics of actinorhodin biosynthesis by Streptomyces
coelicoloer A3(2)”, Journal of Genetic Microbiology (1979), Vol. 114, p.
119-128 |
|
Watanabe A.
and Ebizuka Y.” Unprecedented Mechanism for Chain Length Determination in
Fungal Aromatic Polyketide Synthases”, Chemistry and Biology (2004) Vol.
11, pp. 1101-1106A. |
|
